Blog
FDA & Medical CE Compliance: Ensuring Patient Safety with Aligning Quality Systems with International Medical Directives
2026年7月7日
Introduction: The Regulatory Imperative for Aesthetic Device Excellence
In the rapidly evolving field of medical aesthetics, the integration of advanced technologies such as diode lasers (operating at 755nm, 808nm, and 1064nm wavelengths) is transforming patient care. However, the foundation of clinical efficacy and patient safety lies not just in the technology itself, but in the robust Quality Management System (QMS) that governs its design, manufacture, and deployment. For clinic owners, procurement managers, and medical directors, ensuring that equipment aligns with international medical directives—such as the Medical Device Regulation (MDR) in Europe and FDA requirements in the United States—is non-negotiable. This comprehensive guide explores the critical intersection of quality systems and regulatory compliance, providing the technical and strategic insights necessary to make informed procurement decisions and uphold the highest standards of patient safety .
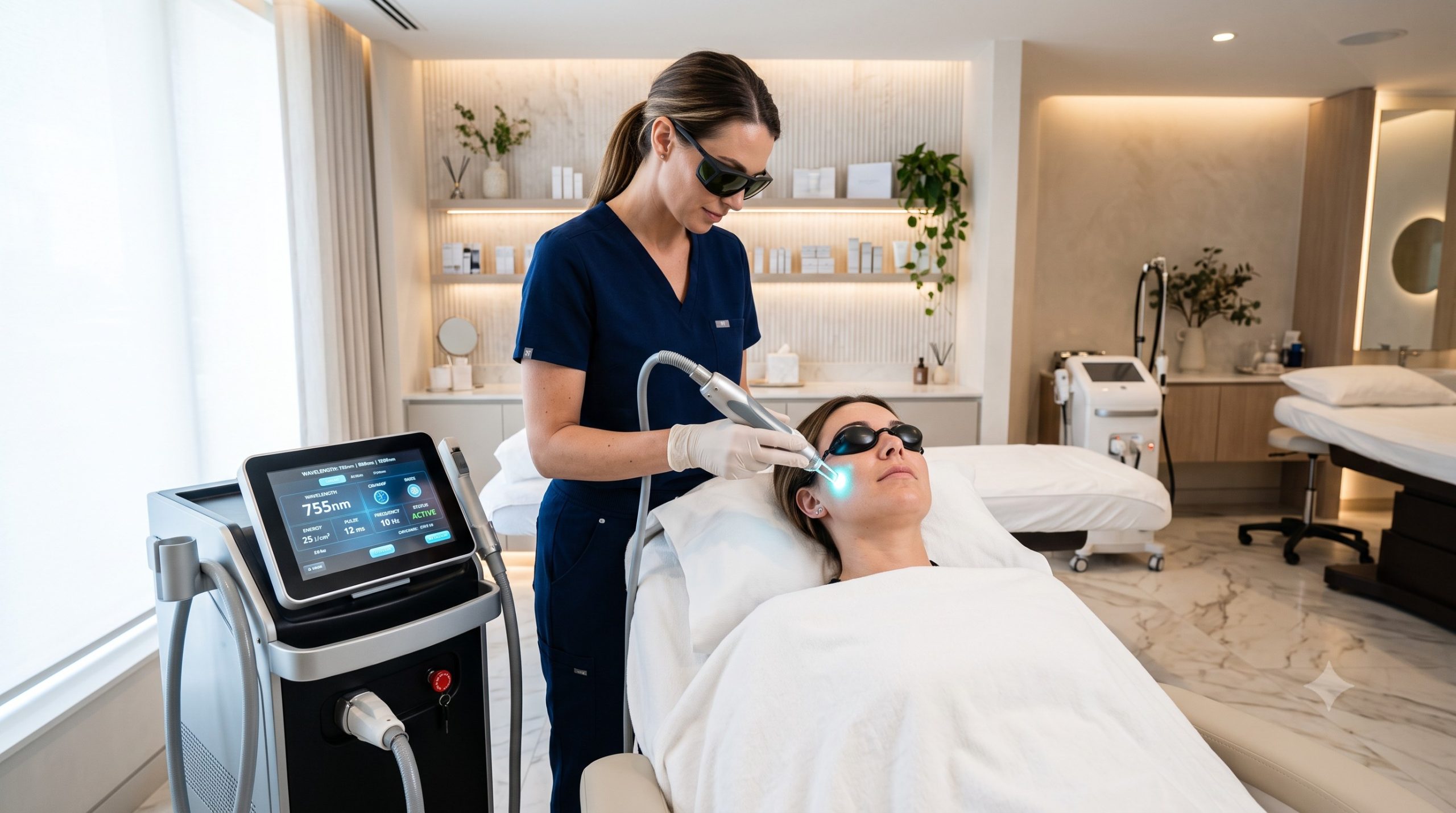
The Global Regulatory Landscape: ISO 13485 as the Cornerstone
At the heart of international medical device compliance is ISO 13485:2016, the internationally recognized standard for quality management systems in the medical device industry. This standard provides a practical foundation for manufacturers to address regulatory responsibilities and demonstrate a commitment to safety and quality . For medical aesthetic devices, adherence to ISO 13485 is often a prerequisite for market access in major jurisdictions. A certified organization proves its ability to consistently provide medical devices that meet customer and applicable regulatory requirements, covering processes from design and development to production, installation, and servicing .
Recent regulatory shifts have underscored the importance of global harmonization. As of February 2, 2026, the U.S. Food and Drug Administration (FDA) has significantly amended its Quality System (QS) Regulation (21 CFR Part 820) to align more closely with ISO 13485, reformatting it as the Quality Management System Regulation (QMSR) . This harmonization is welcomed by the global medtech industry, as it moves towards a single global standard for QMS compliance, reducing the burden of maintaining disparate systems for different markets . For U.S.-focused companies that previously only complied with 21 CFR 820, this means ensuring their QMS aligns with ISO 13485, presenting both a challenge and an opportunity for operational improvement .
Key Differences and the Impact of FDA QMSR Harmonization
The shift from the QS Regulation to the QMSR brings several key changes:
- Risk Management Emphasis: While 21 CFR 820 considered risk in certain aspects like design controls, ISO 13485 has a stronger emphasis on risk management across the entire QMS and device lifecycle. The new QMSR incorporates a risk-based decision process, reflecting the principles of standards like ISO 14971 .
- Documentation and Record Types: The FDA is removing specific requirements for Device Master Record (DMR), Design History File (DHF), and Device History Record (DHR) record types. Instead, product specifications and procedures will now be located in the manufacturer’s Medical Device File (MDF), aligning with ISO 13485’s approach .
- Continuous Improvement: ISO 13485 introduces a clear requirement for the continuous improvement of the QMS’s performance, a concept that is wider than the robust Corrective and Preventive Action (CAPA) system previously required by 21 CFR 820. This makes the QMS a living entity, demanding senior management commitment .
- Customer Satisfaction: A new requirement for 21 CFR 820 is the inclusion of customer satisfaction as part of the QMS, which has always been a key element of ISO 13485 .
CE Marking and the European Union Medical Device Regulation (MDR)
For a medical aesthetic device to be placed on the market within the European Economic Area (EEA), it must bear the CE mark. This marking is a legal declaration that the device conforms to all applicable EU legislation, including the MDR (Regulation (EU) 2017/745), and that its performance, safety, and benefits have been proven for its intended use . This is not a marketing label but a legally binding statement of compliance .
A critical aspect of the MDR is its extension to certain aesthetic products without an intended medical purpose, as listed in Annex XVI. This group includes equipment intended for hair removal, skin resurfacing, and other skin treatments, which now fall under the MDR’s regulatory framework . Manufacturers of these products must comply with classification and CE-marking requirements, including the need for a conformity assessment by a Notified Body for higher-risk classes (like IIa for active hair removal devices) . This ensures that even aesthetic-focused equipment meets rigorous safety and performance standards .
The Role of Notified Bodies and the Conformity Assessment Process
For most medical devices, the CE marking process involves a Notified Body—a private organization designated by an EU member state to conduct conformity assessments . The process requires the manufacturer to:
- Establish a QMS compliant with the MDR (based on ISO 13485).
- Prepare comprehensive technical documentation (Annex II of the MDR) describing the device, its design, manufacturing, and risk management solutions.
- Conduct a clinical evaluation to demonstrate safety and performance.
- Have the QMS and technical documentation audited by the Notified Body.
- Issue an EU Declaration of Conformity .
This rigorous process, which includes regular surveillance audits and unannounced audits for higher-risk devices, ensures that the device continues to meet safety and performance standards throughout its lifecycle .
| Key Parameter | Technical Specification (Example Value) | Clinical Impact |
|---|---|---|
| Wavelength / Laser Type | 755nm / 808nm / 1064nm Diode | Determines depth of penetration and chromophore (e.g., melanin) targeting, critical for treating different Fitzpatrick skin types. |
| Energy Density (Fluence) | Up to 30 J/cm² (Adjustable) | Sufficient energy required to achieve target temperature for Selective Photothermolysis; must be balanced with cooling for safety. |
| Pulse Width | 10ms – 300ms (Adjustable) | Shorter pulse widths target smaller structures (e.g., hair follicle) to confine thermal damage, reducing side effects. |
| Cooling System | Advanced Sapphire Contact Cooling | Actively cools epidermis to prevent burns, reduces pain, enables higher fluence, and increases patient comfort. |
| Spot Size | Up to 20mm x 20mm | Larger spot sizes enable deeper tissue penetration and faster treatment times, improving clinical throughput. |
Technical Specifications and Clinical Safety Metrics
While regulatory compliance is the bedrock, the clinical success and safety of a medical aesthetic device are defined by its technical performance parameters. When evaluating a system, clinicians and procurement specialists must scrutinize the specifications that govern tissue interaction.
Core Technical Parameters
- Wavelength (e.g., 755nm, 808nm, 1064nm): The wavelength determines the depth of penetration and the primary chromophore targeted. For hair removal, 755nm is optimal for lighter skin, 808nm is a versatile ‘gold standard’ for a wide range of skin types, and 1064nm is preferred for deeper penetration and safer treatment of darker skin (Fitzpatrick IV-VI) .
- Energy Density (Fluence): Measured in J/cm², this is the energy delivered per unit area. It is a critical factor for achieving the desired thermal effect on the target (e.g., hair follicle) while minimizing damage to surrounding tissue. The optimal fluence must be balanced with pulse duration and cooling .
- Pulse Width (Duration): Measured in milliseconds (ms), this is the duration of the laser pulse. The principle of Selective Photothermolysis dictates that the pulse width must be shorter than or equal to the target’s Thermal Relaxation Time (TRT) to confine the thermal damage to the target, preventing collateral damage .
- Spot Size: A larger spot size allows for deeper penetration of the laser energy and faster treatment sessions. It is a key factor in clinical throughput and patient comfort .
Cooling Systems and Patient Safety
Protecting the epidermis is paramount during aesthetic treatments. Advanced systems integrate sophisticated cooling mechanisms, such as Sapphire Contact Cooling, to actively cool the skin surface before, during, and after the laser pulse. This approach minimizes the risk of burns, reduces pain, and allows for the use of higher, more effective fluences, thereby enhancing both safety and efficacy .
Clinic Audit Protection and Procurement Strategy
For clinics, investing in a device that aligns with international directives is a critical business and safety decision. A device with a valid CE mark (including the four-digit Notified Body number) and compliance with FDA regulations provides audit protection and ensures that the clinic is not operating with unregulated, potentially hazardous equipment . The new FDA QMSR, which incorporates ISO 13485 by reference, underscores the importance of a manufacturer’s commitment to quality and regulatory compliance .
Long-Term Value and Risk Management
Choosing a device from a manufacturer with a certified QMS (ISO 13485) and a robust risk management process (ISO 14971) offers several advantages:
- Consistent Device Performance: A certified QMS ensures that the device’s output (e.g., energy density, pulse width) remains consistent and reliable over its lifetime.
- Traceability and Quality: Rigorous documentation ensures full traceability of components and manufacturing processes, which is crucial in case of a device failure or safety incident.
- Regulatory Confidence: It provides a strong defense during regulatory or insurance audits, demonstrating that the clinic has invested in safe, approved technology.
Conclusion: Prioritizing Compliance for Superior Clinical Outcomes
Aligning quality systems with international medical directives is not merely a regulatory hurdle; it is the foundational principle for ensuring patient safety, achieving superior clinical outcomes, and building a reputable medical aesthetics practice. The convergence of FDA regulations with ISO 13485 and the stringent requirements of the EU MDR signal a global shift towards higher standards of quality and risk management. As a clinic decision-maker, selecting devices from manufacturers that demonstrate a deep commitment to these standards—through certified QMS, rigorous risk management, and a culture of continuous improvement—is the only pathway to sustainable success and patient trust. By prioritizing regulatory compliance and understanding the detailed technical specifications, clinics can ensure they are providing safe, effective, and world-class aesthetic care.
Frequently Asked Questions (FAQ)
What is the difference between FDA ‘clearance’ and FDA ‘approval’?
FDA clearance (via the 510(k) pathway) applies to moderate-risk (Class II) devices that are shown to be substantially equivalent to a device already on the market. It is a less rigorous process than FDA approval, which is reserved for high-risk (Class III) devices that require extensive clinical trials to prove safety and efficacy . For aesthetic lasers, many are classified as Class II and are cleared, not approved. However, the updated QMSR ensures that all devices, regardless of clearance pathway, are manufactured under a quality system aligned with ISO 13485 .
What is a Notified Body and why is its number important on a CE mark?
A Notified Body is an independent organization designated by an EU member state to assess the conformity of certain medical devices before they are placed on the market . When a CE mark is followed by a four-digit number, it indicates that a Notified Body has been involved in the conformity assessment process, which is required for devices in higher risk classes (e.g., Class IIa and above) . The presence of this number provides an extra layer of assurance that a third-party expert has verified the device’s compliance with the MDR .
How does the new FDA QMSR regulation affect my clinic’s purchasing decisions?
The FDA’s shift to the QMSR means that the agency now places even greater emphasis on a manufacturer’s QMS being aligned with ISO 13485 . When evaluating new equipment, clinics should prioritize devices from manufacturers who hold an ISO 13485 certification. This demonstrates that the manufacturer has a robust, internationally-recognized quality management system in place, providing assurance of consistent product quality, safety, and compliance with the latest regulatory expectations .